研究背景
为了在未来实现绿色和可持续的能源社会,人们致力于开发高效的能源存储系统。锂离子充电电池 (LIB) 的广泛成功归因于其高能量密度、长期耐用性以及大量可用主体结构的存在。近年来,人们特别关注提高除了过渡金属阳离子的氧化还原之外,正极材料还通过氧化物阴离子的氧化还原(O−、O22−、O2−和O2的形成)来提高正极材料的容量。
然而,需求不断增长对于续航里程更长的电动汽车,需要开发具有更高体积能量密度的电源。为了满足这些要求,使用不同载流子离子(例如Li+、Na+、Mg2+、Cl–和F–)的各种类型的电池(例如全固态、空气电池型和氧化还原液流型)已被提出。其中,以氟离子作为载体的氟离子电池(FIB)在理论高体积能量密度方面非常有前途。氟的电负性最高,并且具有高的氧化还原电位。F−/F2的氧化还原电位为2.87 V(vs. SHE)(Cl−/Cl2 为 1.36 V,I−/I2为 0.54 V)允许宽电化学电位窗口,从而形成高电压电池。高电化学容量转换型化合物(例如 Cu/CuF2)的单位重量的高电化学容量也是一个优势。
然而,电子电导率的急剧下降和充放电过程中的巨大体积变化严重损害了倍率性能和循环稳定性。由于这些原因,作为转换类型的替代方案,从能够拓扑定向氟插层的材料中寻找候选电极似乎是有希望的,就像LIB的情况一样(例如,LiCoO2和LiCo1/3Ni1/3Mn1/3O2)。
据报道,化合物中的化学拓扑氟化,例如 LaSrMnO4, Sr2MnO3F和Sr3Fe2O5F2,其 Ruddlesden−Popper 钙钛矿结构为 An+1BnO3n+1 (n =1, 2),其中 A 为稀土元素或碱土元素,B是一种过渡金属。尽管与转化型化合物相比,这些化合物表现出相对较高的倍率性能和循环稳定性,但它们的容量为83 mA h/g(每 Sr2MnO3F 约 0.98 F−)和 118 mA h/g (每 Sr3Fe2O5F2 约2F−)比 LIB 中使用的典型正极容量小。
成果简介
京都大学人类与环境研究院Kentaro Yamamoto教授发现化学氟化的La1.2Sr1.8Mn2O7−δF2氟氧化物可以通过充分利用岩盐相中的间隙位点以电化学方式脱嵌(和嵌入)两个氟离子。出乎意料的是,La1.2Sr1.8Mn2O7−δF2 氟氧化物可以进一步局部氟化到 2F以上,达到 200 mA h/g 的容量,从而可以开发具有高能量密度和耐用性的 FIB。
有趣的是,这些过量的氟离子被引入钙钛矿中,形成氧-氧键以进行电荷补偿(即阴离子氧化还原),如锂离子电池和钠离子电池的脱锂/脱钠正极材料中所见。据我们所知,这是第一份关于在阴离子物质电化学嵌入后引入空穴并形成氧-氧键的报告。鉴于钙钛矿化合物的丰富性,这些结果有望促进FIB正极材料的开发,因为具有阴离子氧化还原作用的混合阴离子化合物可以应用于活性材料。
该工作以“Double-Layered Perovskite Oxyfluoride Cathodes with High Capacity Involving O−O Bond Formation for Fluoride-Ion Batteries”为题发表在Journal of the American Chemical Society上。
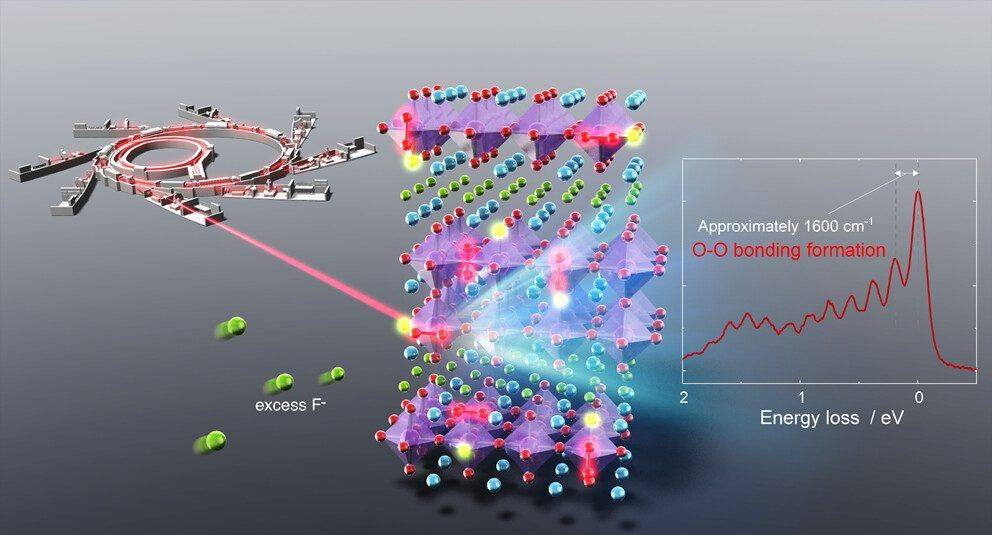
研究亮点
(1) 化学氟化的La1.2Sr1.8Mn2O7−δF2氟氧化物可以实现2F以上的的嵌入脱出,达到 200 mA h/g 的容量。
(2 ) 首次发现除了 0 < x < 2 的常规 Mn 氧化还原外,这种氟氧 化物还可以通过形成 O-O(阴离子氧化还原)将过量的氟离子 (2 < x < 4) 掺入钙钛矿块中。
图文导读
La1.2Sr1.8Mn2O7−δFx (0 ≤ x ≤ 2)氟氧化物岩盐板中的电化学插层
La1.2Sr1.8Mn2O7−δFx (0 ≤ x ≤ 2) 氟氧化物在岩盐板中的电化学插层。采用化学氟化的La1.2Sr1.8Mn2O7−δF2氟氧化物作为起始正极活性材料。根据中子衍射和Rietveld分析的结果,证实化学氟化的La1.2Sr1.8Mn2O7−δF2氟氧化物中形成了氧缺陷(δ∼0.46)。通过球磨 La1.2Sr1.8Mn2O7−δF2 氟氧化物与 La0.9Ba0.1F2.9 和 VGCF 制备正极混合物,如 FIB 中常用的那样并使用该混合物组装了一个三电极电池,其中铅丝作为参比电极,将其尖端插入固体电解质中,不与各电极层接触。图 1a 显示了 10 mA/g 下 -1.5 至 2.0 V 电压范围内的放电-充电曲线。首次放电时,容量达到 91.2 mA h/g,相当于两个氟离子的容量 (91.7 mA h/g),表明氟离子几乎完全从 La1.2Sr1.8Mn2O7−δF2 氟氧化物中脱出。随后的充电过程产生了 86.8 mA h/g 的容量,对应于 95.2% 的放电-充电效率。通过异位 XRD 探测了放电-充电反应过程中正极材料的结构变化(图 1b)。放电时,原始的 La1.2Sr1.8Mn2O7−δF2 氟氧化物 (I4/mmm) 变为 La1.2Sr1.8Mn2O7−δ (I4/ mmm;a = 3.842(2) Å,c = 20.25(2) Å),充电时转变为原始 La1.2Sr1.8Mn2O7−δF2 (a = 3.781(34) Å, c = 23.32(21) Å)。La0.9Ba0.1F2.9 中的衍射峰没有观察到变化。La1.2Sr1.8Mn2O7−δF2 氟氧化物正极放电和1次循环后的 HAADF−STEM 图像(图 1c)和 SAED 图案与所提出的结构一致。这些观察结
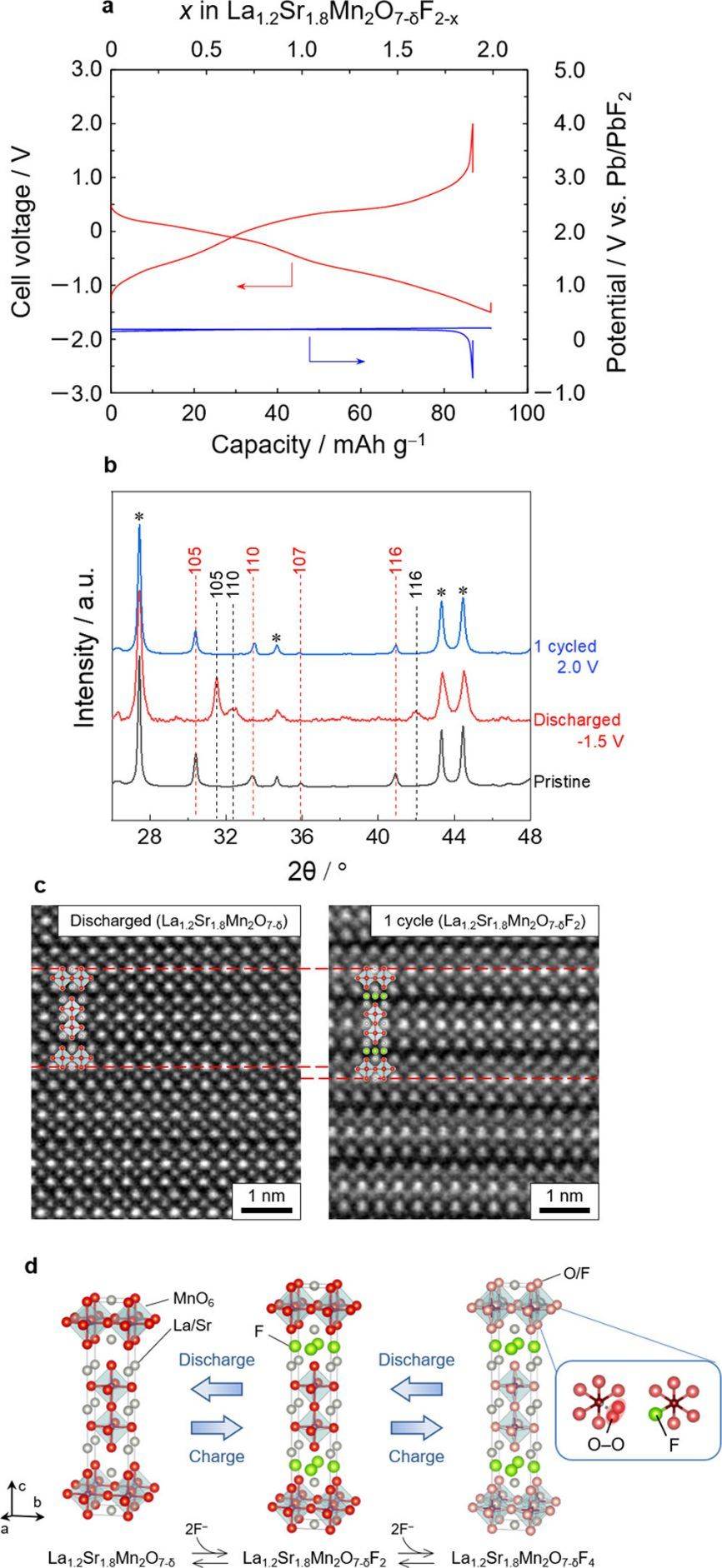
图 1. La1.2Sr1.8Mn2O7−δF2 氟氧化物的电化学性能以及放电/充电过程后的结构变化。a,140 °C、10 mA/g速率下的放电/充电曲线,其中红线和蓝线分别表示电池电压和对电极电位。(b) 含有 La1.2Sr1.8Mn2O7−δF2 氟氧化物的正极复合材料在恒电流放电/充电之前和之后的XRD图。红色和黑色虚线分别是La1.2Sr1.8Mn2O7−δF2氟氧化物和La1.2Sr1.8Mn2O7的峰。星号标记La0.9Ba0.1F2.9(固体电解质)。(c) 沿[010]观察放电和1个周期后La1.2Sr1.8Mn2O7−δF2氟氧化物的高角度环形暗场STEM图像。(d) La1.2Sr1.8Mn2O7−δF2 氟氧化物的放电/充电方案。目前尚不清楚带电的 La1.2Sr1.8Mn2O7−δF2 氟氧化物中 O−O 键和过量氟离子的位置。
果验证了La1.2Sr1.8Mn2O7−δFx 氟氧化物 (0 ≤ x ≤ 2) 中的可逆电化学嵌入和脱出(图 1d)。原子分辨率 STEM−EELS 绘图支持氟离子的插入/提取,其中氟离子占据岩盐板的间隙位置。此外,电极La1.2Sr1.8Mn2O7−δF2氟氧化物表现出优异的循环稳定性和倍率性能。
La1.2Sr1.8Mn2O7−δFx 氟氧化物 (0 ≤ x ≤ 4) 的电化学进一步氟化
到目前为止,我们已经证明了La1.2Sr1.8Mn2O7−δFx氟氧化物(0≤x≤2)优异的电化学反应性能。尽管 La1.2Sr1.8Mn2O7−δF2 氟氧化物是通过化学氟化获得的终端相,最近对 LIB 氧化物的研究利用氧氧化还原化学来实现扩展容量(例如,Li2Ru0.75Sn0.25O和 Li1.3Nb0.3Mn0.4O2)。
考虑到这一点,我们尝试通过将上限截止电压从 2.0 V 提高到进一步氟化(图 2)。当电化学电池在首次放电后充电至 3.0 V(容量相当于两个氟离子)时,在 2.0 V 左右出现宽阔的平台,总充电容量达到约 250 mA h/g(图 2a)。随后的放电过程提供了190mAh/g的大容量,是初始放电过程容量的两倍。
这种过量的电化学氟化也是局部的;3.0 V 充电的 La1.2Sr1.8Mn2O7−δF2 氟氧化物正极的 XRD 图案与原始正极相似(图 2b),并且这些正极的 ADF 图像显示规则的阳离子排列(图 2c)。此外,3.0 V 带电正极中的原子分辨率 EELS 映射(图 2c)显示,除了岩盐板中的间隙位点之外,钙钛矿块中还存在过量的氟离子。3.0 V 充电正极的 XRD 图案与原始 La1.2Sr1.8Mn2O7−δF2 氟氧化物
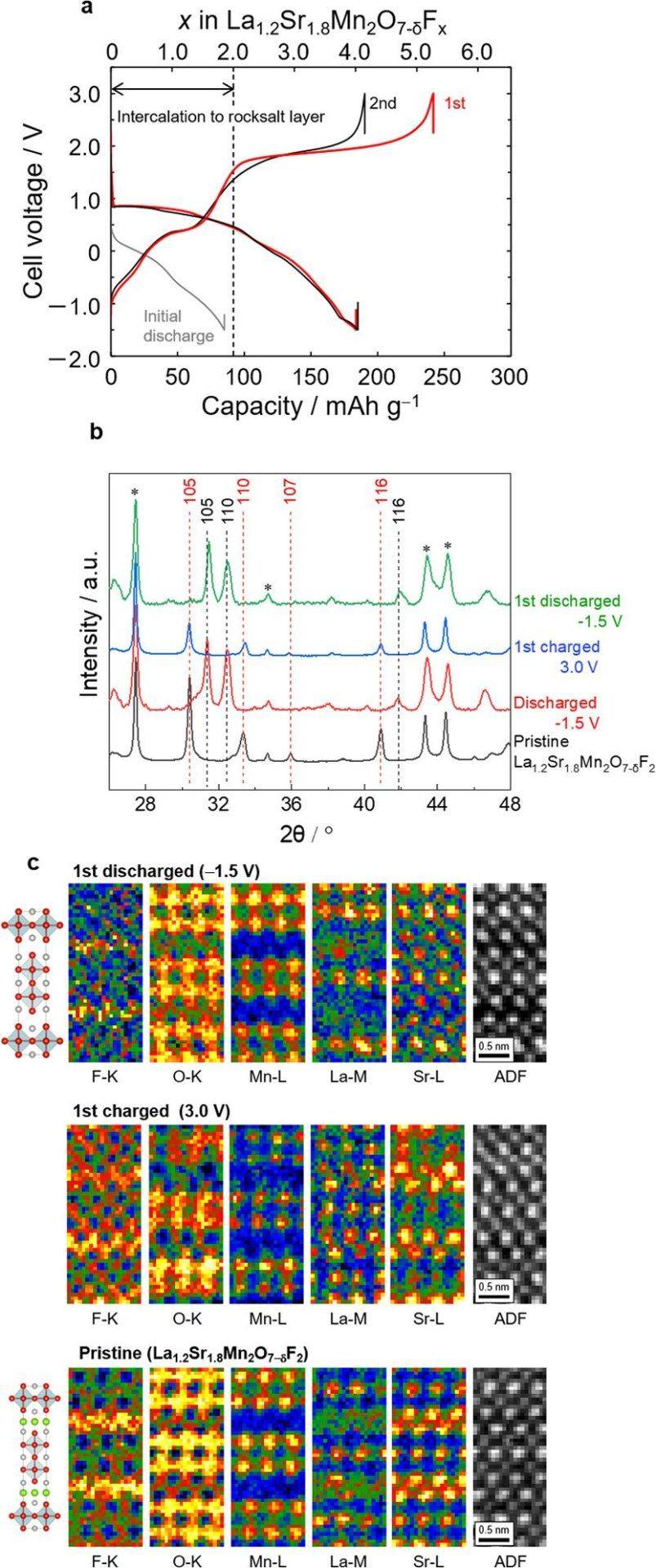
图 2. La1.2Sr1.8Mn2O7−δF2 氟氧化物在较高电压下充电时的充放电特性和结构变化。(a) 在 140 °C 下以 10 mA/g 的速率充电/放电曲线,其中 PbF2/AB 复合材料包含在固体电解质和作为氟源的 Pb 箔之间的阳极侧。(b) 含有 La1.2Sr1.8Mn2O7−δF2 氟氧化物的阴极复合材料在恒电流充电/放电之前和之后的 XRD 图。红色和黑色虚线对应于La1.2Sr1.8Mn2O7−δF2氟氧化物和La1.2Sr1.8Mn2O7。星号表示La0.9Ba0.1F2.9电解质。请注意,为了获得 La1.2Sr1.8Mn2O7−δF2 氟氧化物更清晰的衍射峰,我们使用了活性材料含量较高(~60 wt%)的阴极复合材料(图 S10)。(c) 沿[010]恒电流充电/放电前后La1.2Sr1.8Mn2O7−δF2氟氧化物的原子分辨率STEM-EELS映射图像。为简单起见,显示了理想的结构。
的 XRD 图案接近。随后的放电过程产生了脱氟的 La1.2Sr1.8Mn2O7−δ,经 XRD 和 EELS 证实(图 2b,c)。由于钙钛矿基结构固有的鲁棒性,La1.2Sr1.8Mn2O7−δF2氟氧化物的体积。在第一次充电和第一次放电之间可逆地变化,膨胀最多6%,这比报道的FIB的正极材料要小[例如 CuF2 (193%)、BiF3 (49%)、LaSrMnO4F2−x (17%),和 La2CoO4F2−x (13%)。每次循环的容量提供约200mAh/g(四电子反应),充放电的容量可逆性保持接近100%,如图3a所示,除了第一次充放电过程为80%效率。
第一次循环的不可逆充电容量可能归因于多种副反应,包括固体电解质La0.9Ba0.1F2.9的可忽略不计的轻微氧化;VGCF 轻微氟化,通过恒电流测量和 X 射线光电子能谱证实;和部分氧释放,这得到了后面讨论的 Mn L 边X 射线吸收光谱 (XAS) 的支持,如在 LIB 的富锂正极中所见。放电容量随着充放电循环逐渐增加并保持稳定至少30次循环后容量无明显下降,可逆容量达到200mAh/g。30 次循环期间的平均充电和放电电压几乎恒定。
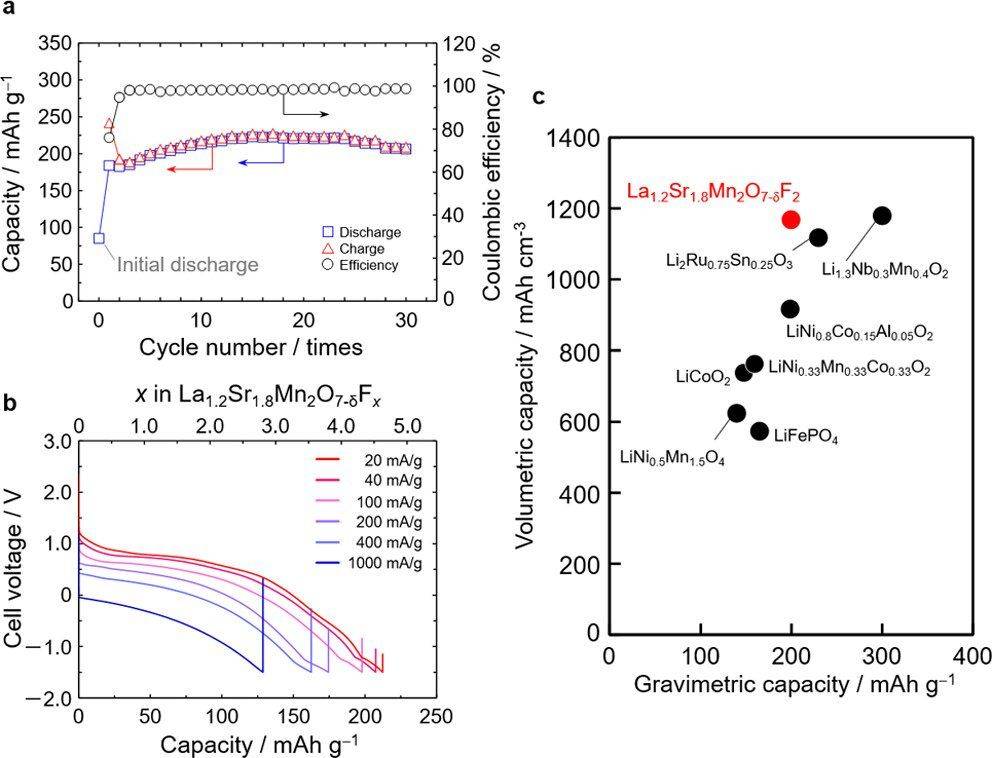
图 3. La1.2Sr1.8Mn2O7−δF2 氟氧化物进一步氟化后的电化学性质。(a)放电/充电容量和效率的循环性能。效率计算如下:第n次放电容量除以第n次充电容量。(b) La1.2Sr1.8Mn2O7−δF2 氟氧化物在不同电流倍率下循环15次后的放电曲线。充电条件相同,上限电压为3.0V,电流率为10mA/g。(c) LIBs 中报告的 La1.2Sr1.8Mn2O7−δF2 氟氧化物和阴极材料的体积/重量容量图
通过 EIS 测量的正极和电解质之间的界面电阻 (Rint, ca) 在循环过程中几乎恒定,表明正极和电解质之间的界面稳定,不会产生副产物。此外,我们的材料的氟含量在 x = 0 和 4 之间变化,具有相对较高的倍率性能(100 mA/g 时为 200 mA h/g,图 3b),这明显优于之前的研究,例如 Sr2MnO3F37(17 mA h/g(100 mA/g 时)和 Sr3Fe2O5F2(75 mA h/g(100 mA/g 时))。
通过恒电流间歇滴定技术估计的 La1.2Sr1.8Mn2O7−δF2 氟氧化物的氟离子表观扩散系数 (DF−) 为 10−13 至 10−11 cm2/s,与锂的表观扩散系数相当LiCoO2 离子(10−12 至 10−11 cm2/s)。因此,La1.2Sr1.8Mn2O7−δF2 氟氧化物的高倍率性能可能与氟离子在本体中的快速扩散有关。如图 3c 所示,获得的容量远高于典型的 LIB 正极材料,并且就体积能量密度而言,与最近研究的使用阴离子氧化还原的活性材料相当。以La1.2Sr1.8Mn2O7−δF2氟氧化物正极、La2.9Ba0.1F2.9和La箔分别作为正极、固体电解质和阳极进行电化学性能测试。
尽管全电池测试的电流密度低于半电池测试的电流密度,但全电池表现出与半电池相似的充放电曲线形状。由于半电池和全电池的正极复合电极的条件完全相同,因此由于缺乏制造条件的优化,La箔阳极可能导致相对较小的电流密度(即相对较大的极化)。尽管制造条件需要改进,但基于活性材料,全电池的重量能量密度为365 W h kg−1,接近非典型LiCoO2/石墨电池(432 W h kg−1),并且2156 W h L−1 的体积能量密度要大得多(与 1550 W h L−1 相比)。
阴离子氧化还原反应
(脱)氟化的电荷补偿机制通过同步加速器硬/软 XAS 进行了检查(图 4)。而 La L 和 Sr K吸收边没有变化,Mn K 吸收边的能量在氟离子插入的早期阶段(x < 2)转移到更高的能量,但在后期保持不变(2 < x < 4)。从前边缘峰值处的能量变化获得了类似的结果。第一配位的 Mn K 边的 EXAFS 分析显示键长减小到 x = 2.4,然后仅略微减小到 x = 4.8。氟离子插入早期的变化反映了Mn氧化,这与Mn K-edge XANES结果一致。
此外,随着氟离子的插入,第二个最近邻峰的强度降低意味着 Mn−La、Mn−Sr 和 Mn−La、Mn−Sr 的 Debye−Waller 因子增加(即局部畸变增加)。Mn− Mn 键。La1.2Sr1.8Mn2O7−δF2 氟氧化物的 Mn L 边光谱(图 4a)从 x = 0 到 x = 2.0 移动到更高的能量,但不是从 x = 2.0 到 x = 4.8,表明 Mn 离子从 Mn3+ 氧化为前期过程中Mn4+,后期过程中仍保留Mn4+。
放电后,与 x = 0 的样品相比,Mn Ledge 谱(放电)转向较低能量,表明 Mn 离子在第一次充电/放电过程中因部分氧释放而减少。在 O K 边光谱中,在充电前 (x ∼ 0) 530 eV 附近观察到了由 O 1s 能级到 Mn 3d 和 O 2p 轨道杂化态的跃迁所致的国外峰值。529 eV 处的强度在氟离子插入的早期阶段增加 (x < 2)(图 4b),这是通过 Mn3+(d4,高自旋)氧化为 Mn4+(d3)的晶体场稳定能来解释的。)。在 Mn L 边光谱中也可以看到相同的行为(图 4a)。
另一方面,进一步超出x∼2的氟离子插入导致在530.8 eV附近出现新的O K边缘峰,并且该峰的强度随着x的增加而增加,而在氟离子萃取时它可逆地消失。O K 边缘峰的增加和 2 < x < 4 时锰的氧化未发生变化,强烈表明氧化的氧物质参与电荷补偿,正如在富锂正极中观察到的那样。当氟离子插入超出 x > 2 时,F Kedge 光谱(图 4c)的前边缘区域表现出一个小峰,可归因于金属-氟键,其强度随着 x 的增加而增加,表明形成了 Mn−钙钛矿块中的 F 键得到 STEM-EELS 的支持(图 2c)。
为了研究Mn 3d− F 2p杂化轨道中空穴形成的可能性,我们通过第一性原理计算计算了LaSr2Mn2O7F2的部分态密度(pDOS),该LaSr2Mn2O7F2近似于La1.2Sr1.8Mn2O7−δF2氟氧化物,并且发现只有 Mn 3d 和 O 2p 轨道存在于费米能级附近,与这些轨道相比,F 2p 的能量非常低。
这些结果表明,当过量的氟离子插入正极时,F2p 轨道中不会发生空穴形成;FK-edge前边的变化可能是由于O 2p 轨道中形成空穴,改变了邻近Mn 的3d 轨道,进而改变了与Mn 相邻的F 2p 轨道。为了通过实验确认带电 La1.2Sr1.8Mn2O7−δFx 氟氧化物正极中氟的电子结构,将 K 边缘 XANES 和氟发射光谱测量与 RIXS 测量相结合是有用的。
然而,在这个系统中,很难由于复合电极上混合了 La0.9Ba0.1F2.9 固体电解质,因此从上述测量中提取信息。为了进一步阐明氧化氧的性质,在真空中进行了 O K 边 XAS 光谱中 528.5 至 533.0 eV 的 RIXS 测量(图 S24)。获得的 530.8 eV 处的 RIXS 谱(图 4d)在弹性线周围从 5 到 0 eV 处表现出离散的能量损失峰,代表基态势能表面的几个振动能级。第一振动能级的频率为 1591 cm−1,类似于分子 O2(约 1600 cm−1),最近在充电过程中在充电的 Na0.75[Li0.25Mn0.75]O2 和 Li1.2Ni0.13Co0.13Mn0.54O2 中观察到这种分子真空下的 RIXS。
由于本研究中使用无机 La0.9Ba0.1F2.9 作为电解质,因此正极和有机电解质之间的副反应不能形成 O−O 物种。1-2 eV中的附加特征范围不是由于 PDOS 的存在,而是由寿命振动干扰引起的振动进程的强度调制引起的。该结果表明在带电的 La1.2Sr1.8Mn2O7−δF2 氟氧化物 中形成了 O−O 键。放电后,这种振动消失,这意味着 O−O 键的形成/断裂是可逆的。
看来 O−O 键的形成不仅可以解释电荷补偿(阴离子氧化还原),而且还可以产生额外的空位来填充多余的氟离子。假设 O−O 键的形成符合 RIXS 结果的预期,两个电子的电荷补偿会增加 0.5 个空位位点 (O2− → 1/2O2 + 2e−),并且在不规则阴离子位点上应占据大量氟离子。我们尝试使用 ABF-STEM 显微分析来确定 O−O 键形成过程中带电 La1.2Sr1.8Mn2O7−δF2 氟氧化物正极的结构。未能获得确定结构的清晰图像可能是因为结构高度扭曲。恒电流间歇滴定技术表明,x > 2 时开路电压的极化和迟滞大于 0< x < 2,并且 x > 2 时较大的极化可能是由氧-氧键的形成引起的。可能涉及局部扭曲。在化合物中观察到类似的不可忽略的极化,包括与钠离子电化学脱嵌相关的氧分子的形成。
完全充电的 La1.2Sr1.8Mn2O7−δF2 氟氧化物的 XRD 图案与原始 La1.2Sr1.8Mn2O7−δF2 氟氧化物相比几乎没有变化(图2b),这可能是由于以下两个原因。其一是由于氟离子插入位点已从岩盐相中的二维晶格位置转变为与钙钛矿层中 O−O 键形成位点相关的随机位点。据报道,对于锂离子电池,与锂离子嵌入和脱嵌到岩盐结构中的随机渗滤位点相关的晶格体积膨胀相比,锂离子嵌入和脱嵌到岩盐结构中的随机渗滤位点时,晶格体积变化非常小。
三维层状岩盐氧化物正极[例如层状LiCoO2 (3.6%)、阳离子无序岩盐Li1.25Nb0.25V0.5O2氧化物(1%)、和Li1.25V0.55Nb0.2O1.9F0.1氟氧化物(0.7 %)].这是由于与二维位点的插入/拔出相关的较大的库仑吸引力/排斥力。另一个原因是由于氧的2p轨道引入了空穴,即氧离子的离子半径随着氧化而变小。如上所述,虽然带电的 La1.2Sr1.8Mn2O7-δF2 氟氧化物中 O−O 键是如何形成的以及过量的氟离子如何存在尚不清楚,但这些结果表明 O2 等分子和过量的氟离子可能存在于某些结构中的空腔(如在富锂正极材料或 N2 插层 WO3,中发现的)以及分子状 O2 的形成可能产生了氟离子的不规则位点。
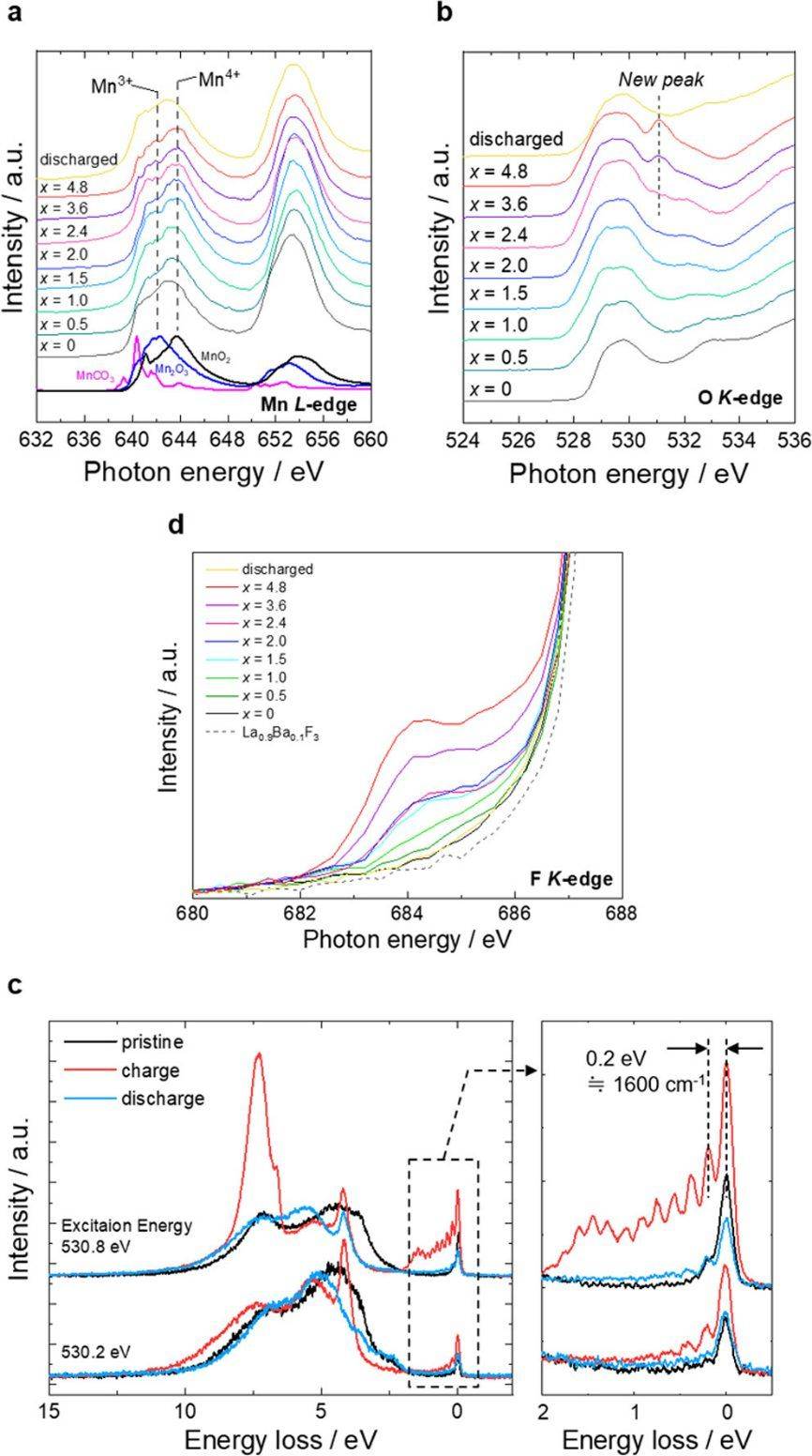
图 4. La1.2Sr1.8Mn2O7−δF2 氟氧化物在第一次充电/放电过程中电子结构的变化。(a,b) 针对充电/放电曲线中不同状态收集的 XAS 光谱变化,如图 2a 所示。(a) Mn L 边吸收和 (b) O K 边吸收。图中的x表示氟化物含量(La1.2Sr1.8Mn2O7−δFx中的x)。(c) F K 边吸收,表明在 x > 2 处形成了Mn−F 键。(d) 在原始带电状态下,在 530.2 和 530.8 eV 的激发能下记录的氧 K 边 XAS 和高分辨率 RIXS 光谱分别为 (3.0 V) 和放电 (-1.5 V) 状态。1600 cm−1 左右的振动频率表明存在 O−O 键。
总结与展望
使用双层 Ruddlesden−Popper 型钙钛矿氟氧化物 La1.2Sr1.8Mn2O7−δFx 证明了氟化物离子的电化学嵌入具有优异的可逆性、循环性和倍率性能。有趣的是,此外与 0 < x < 2 的传统 Mn 氧化还原相比,这种氟氧化物可以通过形成 O−O(阴离子氧化还原)将过量的氟离子 (2 < x < 4) 结合到钙钛矿块中。尽管La1.2Sr1.8Mn2O7−δFx通过形成O−O键而表现出高容量,但其容量仍然与富锂正极相当。
因此,利用相同的反应机理开发比La1.2Sr1.8Mn2O7−δFx具有更高容量的新型正极。需要强调的是,与LIB一样,目前的FIB在循环稳定性、倍率特性、氟离子扩散等电化学性能方面还有进一步改进的空间;可用的策略包括用其他过渡金属进行化学取代、氧空位控制、电极混合物成分的优化、调整颗粒形态以及控制电极和电解质之间的界面接触。这次在140℃下测量电池性能主要是因为所使用的固体电解质的氟离子电导率较低。
因此,开发具有高离子电导率和电化学窗口的新型固体电解质是必要的。鉴于传统钙钛矿材料已知的各种结构和成分,通过形成O−O键捕获过量氟离子的能力为钙钛矿工程增加了新的维度,不仅适用于电池研究,也适用于其他学科。
事实上,具有“密堆积”结构的钙钛矿 ABO3 中分子状 O2 的形成并不明显,总体上可能是一个有趣的研究课题。此外,这项研究充分提高了人们的认识,即电化学是获得新型氟氧化物的强大工具,更广泛地说,是获得可能对各种物理和化学功能产生影响的混合阴离子化合物。
文献链接:
Miki H, Yamamoto K, Nakaki H, et al. Double-Layered Perovskite Oxyfluoride Cathodes with High Capacity Involving O–O Bond Formation for Fluoride-Ion Batteries[J]. Journal of the American Chemical Society, 2024.

免责声明
本文仅代表作者观点,不代表本站立场,著作权归作者所有;作者投稿可能会经本站编辑修改或补充;本网站为服务于中国中小企业的公益性网站,部分文章来源于网络,百业信息网发布此文仅为传递信息,不代表百业信息网赞同其观点,不对内容真实性负责,仅供用户参考之用,不构成任何投资、使用建议。请读者自行核实真实性,以及可能存在的风险,任何后果均由读者自行承担。如广大用户朋友,发现稿件存在不实报道,欢迎读者反馈、纠正、举报问题;如有侵权,请反馈联系删除。(反馈入口)
本文链接:https://www.byxxw.com/zixun/32517.html- 上一篇: 对子莲怎么养?
- 下一篇: 小伙试吃网红“毛巾蛋糕”,撕开包装后,发现事情并不简单